z-Matrix and Internal Coordinates
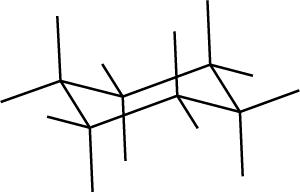
************************************************************ ** Setting up the cyclohexane structure using a z-matrix ****** The System ******************************************* spectrometer(MHz) * spinning_freq(kHz) * channels H1 nuclei H1 atomic_coords * cs_isotropic * csa_parameters * j_coupling * quadrupole * dip_switchboard * csa_switchboard * exchange_nuclei * bond_len_nuclei (1 7) bond_ang_nuclei (1 2 3) tors_ang_nuclei (1 2 3 4) groups_nuclei * ******* Pulse Sequence **** CHN 1 timing(usec) 0 power(kHz) 0 phase(deg) 0 freq_offs(kHz) 0 ****** Variables ********** rCC=1.5 rCH=1 theta=109.4712 ******* Options *********** rho0 I1x observables I1x EulerAngles * n_gamma * line_broaden(Hz) * zerofill * FFT_dimensions * options -zxmatzxmat.dat -s -expcm * The atomic coordinates of the structure created via a z-matrix or internal variables can be inspected with the -s option * The coordinates can be used to draw the strucure in molecular/crystal graphics programs (e.g., CrystalMaker, see the cyclohexane.pdf file) * -s -expcm: export coordinates in a format readable by Crystal Maker * The internal coordinates r_1, phi_1, and theta_1 are defined at the bond_len_nuclei, bond_ang_nuclei, and tors_ang_nuclei lines but they are not set in the Variables section. So, they are only measured, and can be inspected (in the -s option printout) to verify that the molecule was generated correctly
zxmat.dat
C13 1 C13 2 1 rCC C13 3 2 rCC 1 theta C13 4 3 rCC 2 theta 1 60 C13 5 4 rCC 3 theta 2 -60 C13 6 5 rCC 4 theta 3 60 H1 7 1 rCH 2 theta 3 60 H1 8 1 rCH 2 theta 3 180 H1 9 2 rCH 3 theta 4 -60 H1 10 2 rCH 3 theta 4 180 H1 11 3 rCH 4 theta 5 60 H1 12 3 rCH 4 theta 5 180 H1 13 4 rCH 5 theta 6 -60 H1 14 4 rCH 5 theta 6 180 H1 15 5 rCH 6 theta 1 60 H1 16 5 rCH 6 theta 1 180 H1 17 6 rCH 1 theta 2 -60 H1 18 6 rCH 1 theta 2 180